An advanced example
In this part, we will use an example of a protein to illustrate how to conduct MD simulations in real-world research.
Building Model
Again, we use the BO approximation to construct models for proteins. These models include information on bond types, atom types, and force field parameters for various interactions. We usually need the following files:
topology (
topol.topfor example): Contains molecular bonding information, molecular type information, and atomic type informationforce field (
forcefield.itpfor example): contains chemical bond equilibrium positions, chemical bond force constants, non-bond force constants, etc.Commonly used force fields are as follows:
Amber
Charmm
Gromos
OPLS
So far there is no unified database or maintenance of methods for force fields, and each force field is maintained by each company or organization. Developing a force field is a difficult and nuanced process.
Solvent
Unlike systems in vacuum, proteins generally exist in solvents. We therefore need to account for and model the solvent. We can do this in two ways:
Explicit solvent model. It is often used in all-atom simulations, that is, directly introducing solvent molecules into the system. The force field parameters for solvent molecules are usually included in the force field file. Commonly used models for water are SCP, TIP3P, TIP4P, etc.
Advantages: Similar to the actual physical process, the results are more accurate. It can describe solvent-involved processes (e.g. protein-ligand binding). It can also explicitly describe solvent effects such as hydrogen bonding.
Disadvantages: high computational complexity. A system usually contains hundreds or thousands of solvent molecules.
Implicit solvent model. The effect of the solvent on the solute is described by a continuous electric field model. The Generalized Born model is an example of commonly used model.
Advantages: low computational complexity, no need to introduce additional solvent molecules.
Disadvantages: Imprecise, solvent-involved reactions cannot be described.
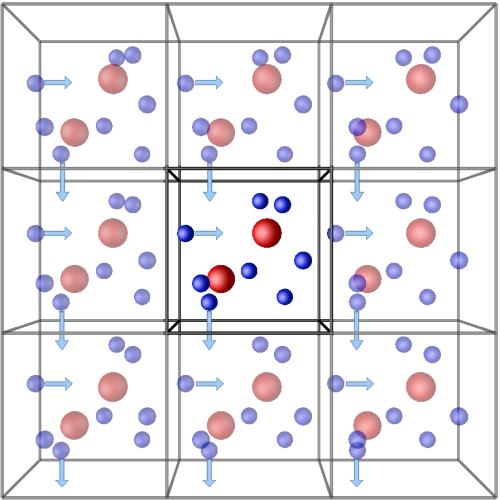
Periodic Boundary Conditions:
Due to limited computation resources it is impossible for us to simulate an infinitely large system, nor to simulate infinite steps. For a protein system, tens of thousands of atoms already require a lot of calculations (the required calculation time is measured in days), but this is still far less than Avogadro’s constant(\(10^{23}\)).
However, simulating a small system will be seriously affected by the interface and cannot reflect the properties of the bulk phase.
To solve this problem, we often use periodic boundary conditions. We confine the system of interest in a box and assume the properties of the actual system can be approximated by an virtual infinite system of repeating side-by-side lattices. If the molecule passes through the box boundary, it will re-enter the box from the opposite boundary, forming a periodic space.
Preparation for simulation
Next, you need to prepare the conformation files of the protein and water molecules. Typically, protein structure files are generated by PDBs, then converted into a format that can be read by molecular dynamics software.
We also need to prepare simulation parameter files, in which you need to set:
temperature
time interval and duration for simulations
the integrator/numerical algorithm, the ensemble for simulations
the temperature/pressure controller
the output-frequency and content of output files
Simulation Commonly used MD simulation software in current research are as follows:
Amber [1]
Commercial software. There is a free version and a high-performance optimized commercial version. The code is closed source and highly engineered. It supports most MD functions and plays an important role in the simulation of protein systems. Amber is often used in simulations of biological systems.
Gromacs [2]
Open-source MD simulation software. The latest release is the Gromacs 2022 release version. With efficient GPU optimization, there is a good developer community for Gromacs. This is mostly used for biological system simulation.
OpenMM [3]
Molecular dynamics simulation software with Python interfaces. Modules are implemented by calling functions from the Python command line, which can directly involve deep learning frameworks such as PyTorch. However, its compatibility has not been fully developed. Mostly used for biological system simulation.
Lammps [4]
MD software for material simulations.
Most MD software is fully optimized on GPU devices, which can provide greater efficiency than CPU devices. Another program, Charmm, can be used for preparing structures for MD but it can not be used for simulations.
In addition, most software is compatible with other formats of force fields; for instance, Gromacs is compatible with Amber or Charmm force fields.